


医学院 新闻动态
2025年5月29日,西湖大学医学院郭天南教授团队,联合广东省人民医院关海霞教授等多个团队,在 EMBO Molecular Medicine 发表了一项基于蛋白质组学的滤泡状甲状腺肿瘤精准分型研究。该研究通过构建并验证一个基于24种蛋白的分类器,有效改善了滤泡状甲状腺腺瘤与癌的鉴别诊断,该技术对甲状腺滤泡肿瘤的术前精准诊断提供了参考信息。
文章链接:
https://www.embopress.org/doi/full/10.1038/s44321-025-00242-2

图1 文章截图
提纲挈领
以下为研究的结果部分详细解读:
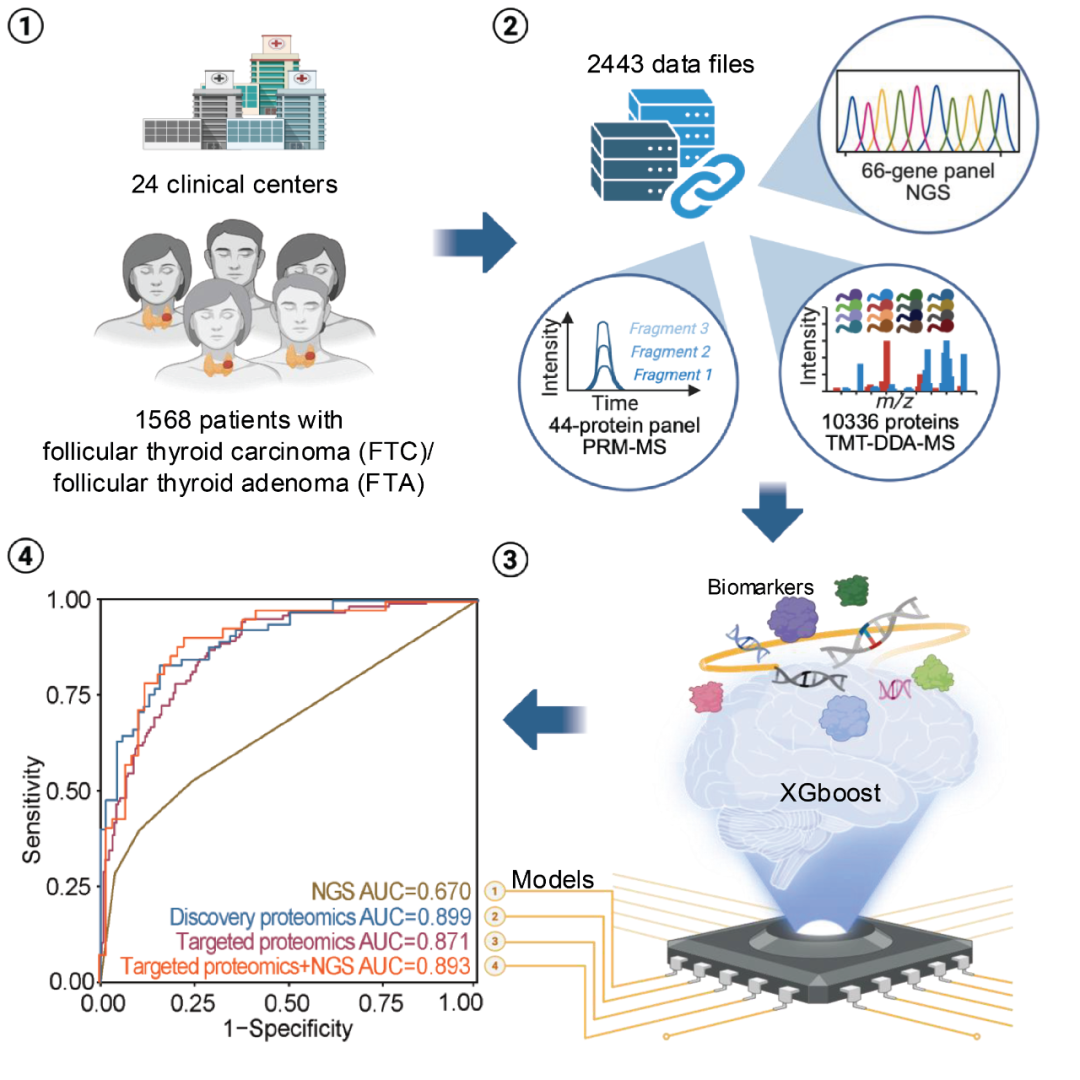
01患者特征与研究设计本研究共纳入来自中国和新加坡24家中心的1568名患者,共收集了2443份样本数据。其中,FTA患者909例,FTC患者659例。患者中位年龄为49岁,女性占比约70%,男女比例为2.4:1。结节中位直径为35 mm,且近半数病例的结节小于40 mm。
研究采用多阶段设计:首先,通过NGS分析609例样本的基因变化;通过TMT标记定量质谱技术分析620例样本的蛋白质表达并进行分类模型构建、比较与优化。随后,在729例样本上实施靶向质谱(PRM)以构建蛋白质分类器,并在三个独立测试集(内部、回顾性、前瞻性)中进行了验证。
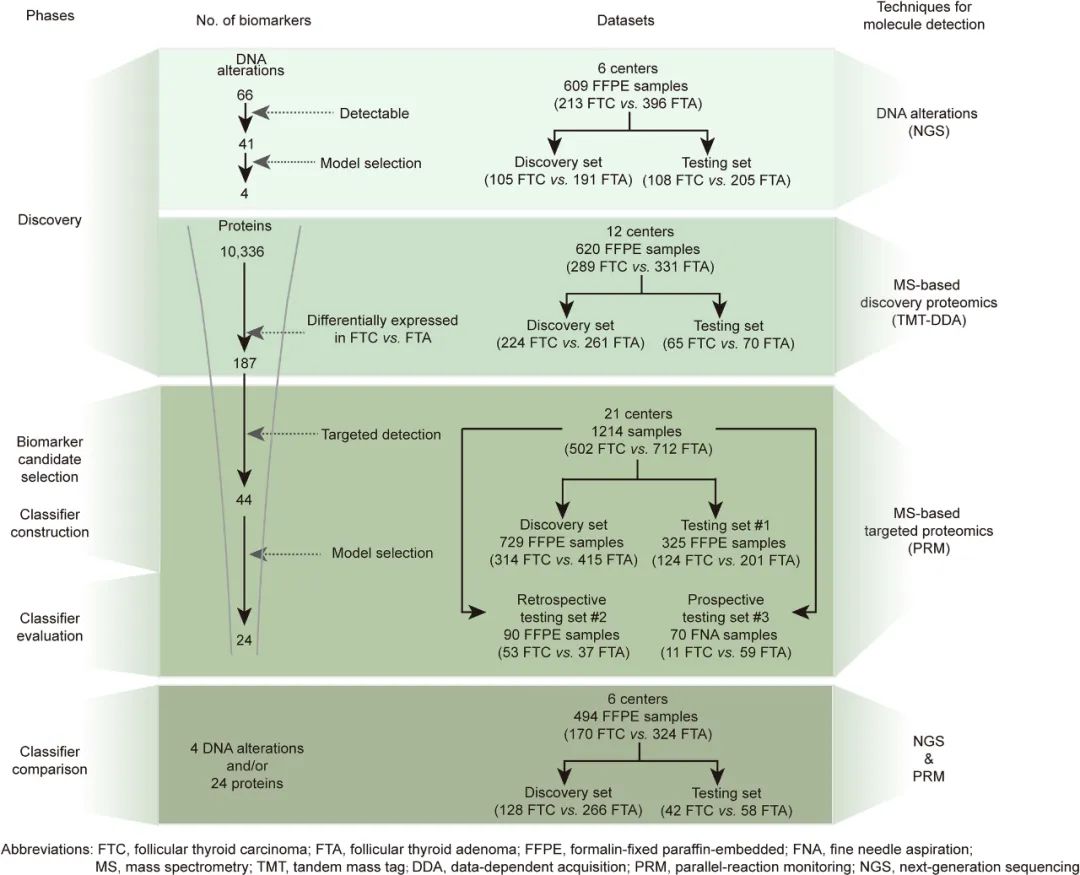
图3 生物标志物发现、分类器开发、性能评估和比较的流程图
02基因组模型不能有效区分FTA与FTC
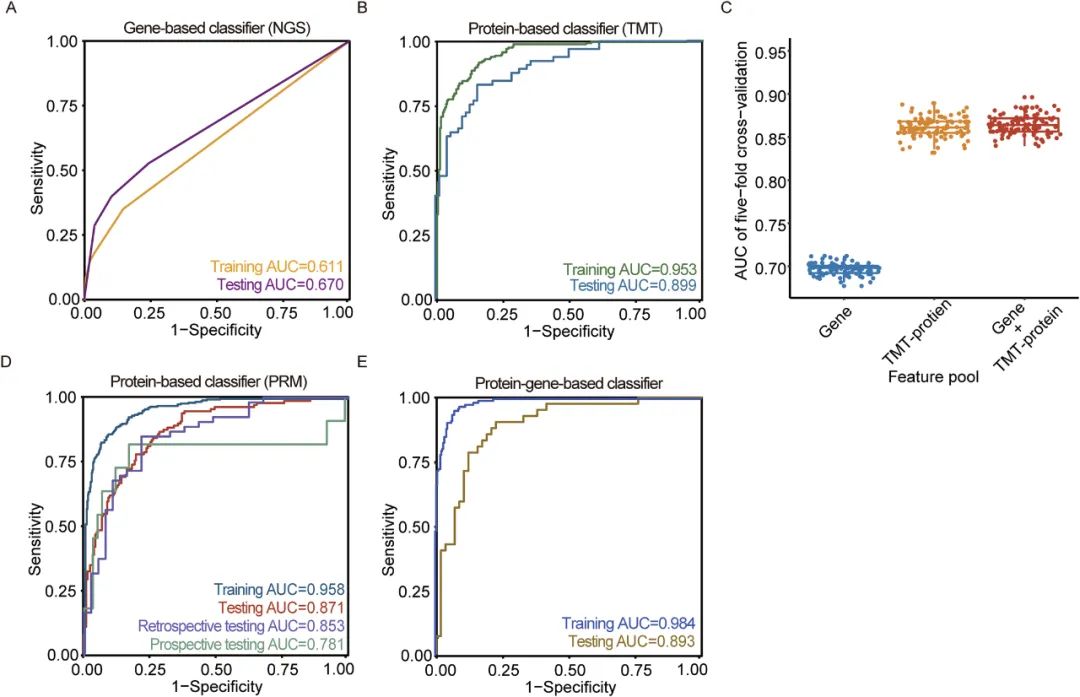
对609例样本进行66基因panel测序发现,仅有41个基因(62.1%)在数据中被检测到。整体突变率为53.4%,其中FTA为46.2%,FTC为66.7%,但46.6%的样本无任何可检突变。
03深度蛋白质组学可显著改善分类性能
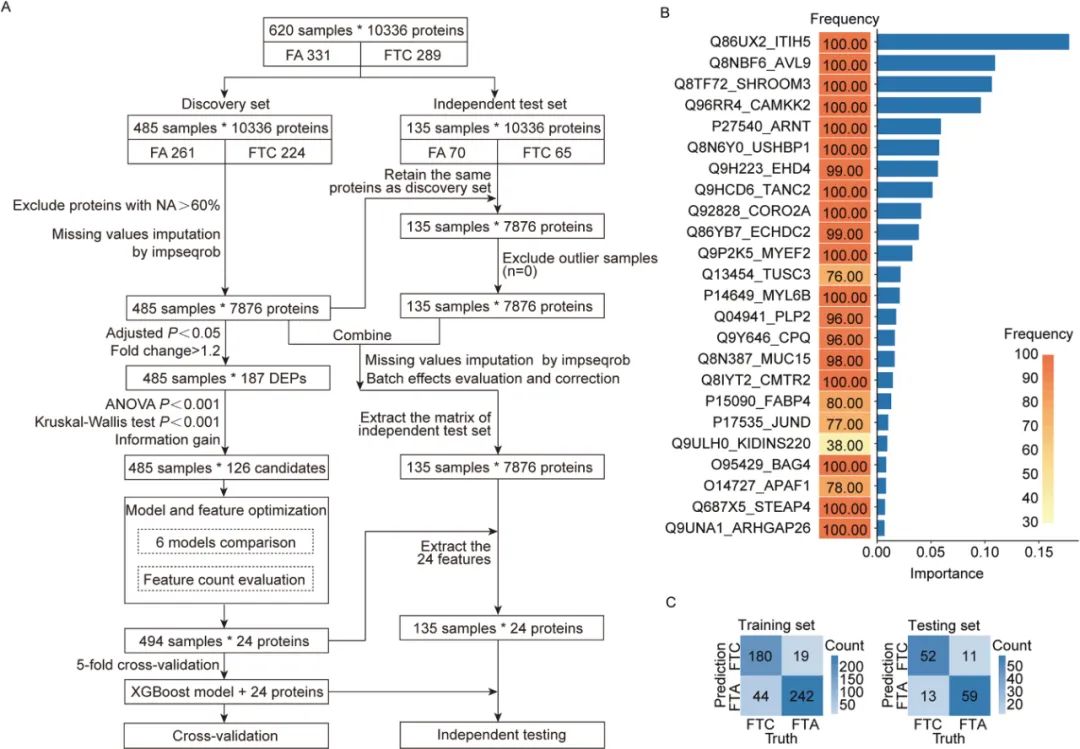
研究通过TMT质谱分析620份回顾性FFPE样本,定量检测到10,336种蛋白,质量控制后用于后续分析的蛋白数为7876。
通过比较,FTC与FTA二者之间差异表达分析识别出187个DEPs,该蛋白群体主要富集于甲状腺激素生成和代谢通路。然而,进一步的降维分析显示,单靠这些DEPs仍难以完全区分FTA与FTC,进一步说明了分子表达水平上的相似性与鉴别二者的困难性。
因此,研究团队利用多种机器学习方法筛选最佳特征数和算法,最终构建了基于24个蛋白的XGBoost模型,在训练、交叉验证和独立测试集中分别获得AUC为0.953、0.905和0.899,显著优于基因模型。同时,该模型在独立测试集中的敏感性、特异性、准确率和NPV均表现良好。进一步分析显示,联合基因数据并未显著提升模型性能,强调蛋白组数据在分类中的主导作用。
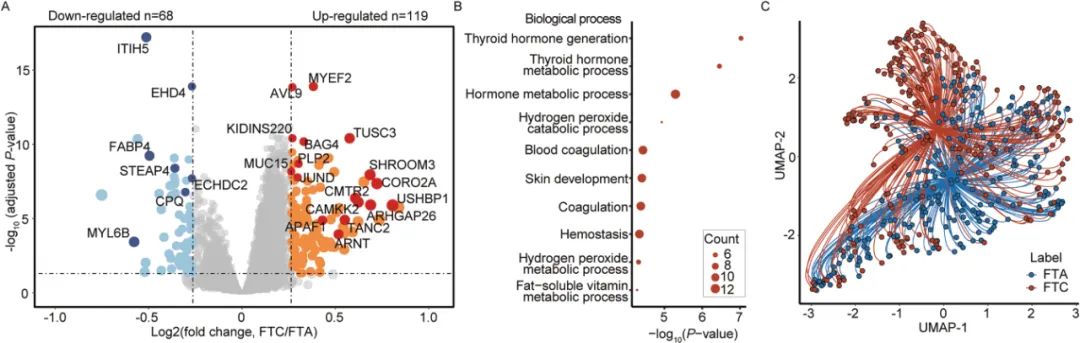
图5 在TMT发现数据集中对FTC与FTA进行的比较蛋白质组学分析
04靶向蛋白质组学模型的开发与验证
考虑到TMT的测试成本高和临床可及性低,研究转向PRM靶向质谱以提升临床实用性。在靶向可以检测到的44个差异蛋白中,筛选出24个蛋白用于构建分类器,并在四个数据集中(总样本1214例)进行测试。
多个中心的外部验证进一步证实了该模型的泛化能力和临床应用前景,尤其对术前诊断具有重要意义。
05基因与蛋白质联合模型的比较分析
在494例同时具备基因和蛋白数据的样本中,研究构建了三个模型:仅基因、仅蛋白,以及二者联合。
结果显示,联合模型AUC为0.893,虽略高于蛋白模型(24蛋白),但提升主要归功于蛋白特征,基因信息的增益有限。该联合模型在独立测试集中表现稳定,准确率为0.820,特异性高达0.897,进一步验证了蛋白质组学在分类性能上的主导性和临床价值。
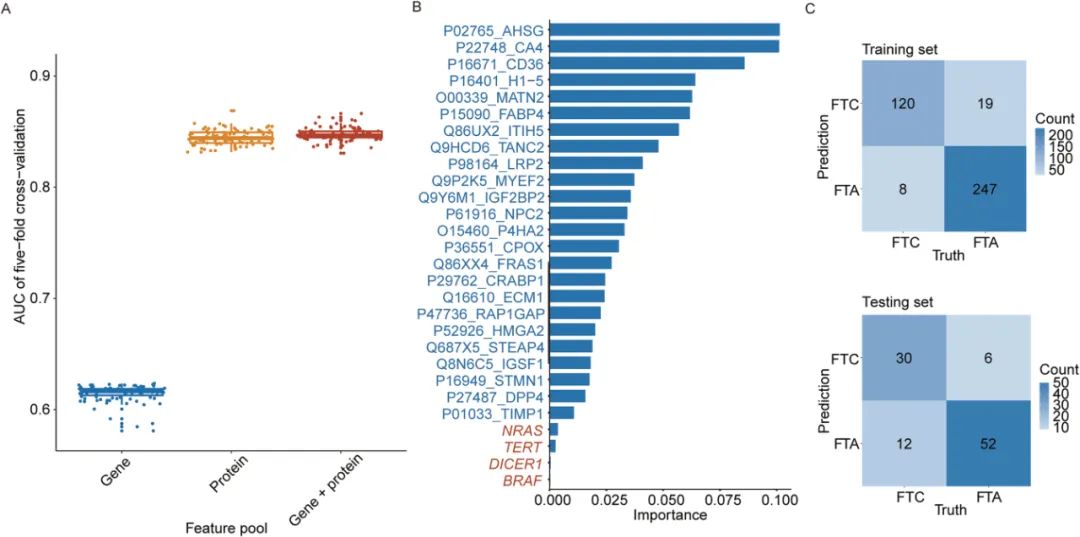
图7 组合特征模型的性能比较及特点
本研究通过整合深度蛋白质组学和靶向蛋白组学,并结合机器学习方法,构建了一个高效、可推广的蛋白质分类器,首次实现了对FTA与FTC的高精度鉴别诊断。
研究不仅为甲状腺结节的精准诊疗提供了新工具,也展示了蛋白质组学在疾病分类与生物标志物开发中的广阔前景。未来,该策略有望拓展至其他组织病理分型难题,为临床病理诊断提供更科学、精准的解决方案。
西湖大学医学院博士后研究员孙耀庭(现为德国马克斯·普朗克生物化学研究所博士后研究员),科研助理王赫(现为新加坡国立大学博士研究生)、访问学生李璐(现为浙江大学博士研究生)等为该研究共同第一作者。西湖大学医学院郭天南教授、广东省人民医院关海霞教授、西湖实验室朱怡研究员为共同通讯作者。
研究得到了慢性非传染性疾病国家科技重大专项、国家重点研发计划、浙江省“尖兵领雁”研发攻关计划、中国博士后科学基金以及医学蛋白质组全国重点实验室自主研究课题资助的支持。感谢西湖大学超级计算机中心提供的数据存储和计算服务。
最新资讯
新闻动态














