


医学院 新闻动态
2026年1月22日,西湖大学医学院郭天南团队在Nature Communications发表最新研究成果,开发了一种名为ProteoAutoNet的共分馏质谱方法,将自动化机械臂与机器学习计算模型相结合,实现了对蛋白质-蛋白质相互作用的高通量、深入的分析,为研究癌症等复杂疾病的分子机制和药靶发现提供了新工具。
文章链接:
https://www.nature.com/articles/s41467-026-68686-9

图1 文章截图
提纲挈领
捕捉蛋白质的“社交网络”
如果把细胞比作一个微观世界的社会,蛋白质就是其中的居民。它们并非孤立工作,而是通过形成复杂的社交网络,即蛋白质-蛋白质相互作用(Protein-Protein Interaction, PPI),用以执行各种生命活动。这个网络的异常,往往是癌症等疾病的根源。
如何系统捕获活细胞中蛋白质-蛋白质相互作用,从而绘制蛋白质的动态“社交网络”图,一直是生命科学领域的重大挑战。共分馏质谱(Co-Fractionation Mass Spectrometry, CF-MS)是一种将天然状态蛋白质复合物进行分馏,并使用质谱分析共流出蛋白质复合物的高通量技术。其核心原理是通过生物化学的分离方法,将细胞或组织提取物中的蛋白质复合物按其物理化学特性,如分子大小或电荷进行分离。在分离过程中,相互作用的蛋白质因共同组成蛋白质复合物而协同迁移,从而在相同馏分中被检测到。同化学交联技术相比,该方法避免过多假阳性。但这种方法高度依赖样品分馏,导致样品制备流程复杂、耗时且劳动强度高。
近年来数据非依赖采集(Data Independent Acquisition, DIA)质谱显著提升蛋白质组定量的通量及稳定性。将DIA与CF-MS结合,有效地推动了CF-MS的发展。然而,复杂的样品前处理仍然是限制其规模化应用的关键因素。例如,人工制备81个CF-DIA-MS可分析的多肽样本需耗时约40小时。即使借助商业自动化平台,处理1920个共分馏样本仍需至少6天。此外,高质量标注数据的稀缺与多样性不足,也进一步制约现有计算方法的预测性能。基于上述挑战,研究团队开发了ProteoAutoNet——一个集成自动化机械臂实验平台与机器学习分析的高效解决方案。
1.自动化机械臂实验平台
1)把CF-DIA-MS做成 “流水线”
ProteoAutoNet的实验模块整合Opentrons自动移液系统与JAKA机械臂,实现CF-DIA-MS样品制备流程的端到端自动化,包括蛋白质复合物分馏后的变性、还原、烷基化、酶解及离线脱盐等关键步骤。该平台将样品处理通量提升约2倍,仅用3天即可完成3个细胞系的9个生物样本(共540个组分)的预处理。
在实验模块中,Opentrons可并行处理最多8块96孔板,并在SDC裂解下完成蛋白质变性、还原与烷基化。单次运行可处理4块孔板(约50到60分钟),酶解步骤单次最多处理6块板(约40分钟)。从已分馏的蛋白质复合物到多肽制备的全流程中,处理4块、8块、12块板分别仅需约16小时、18小时和24小时。
JAKA机械臂支持开源定制的离线脱盐流程。单机械臂可同时处理4块96孔板,4到6块板完成5次脱盐约需4小时,双机械臂配置下,8到12块板约需8小时。通过将自动化流程与必要的人工操作整合,该平台可在2至3天内完成4到12块板从复合物到多肽的全流程处理(图2)。
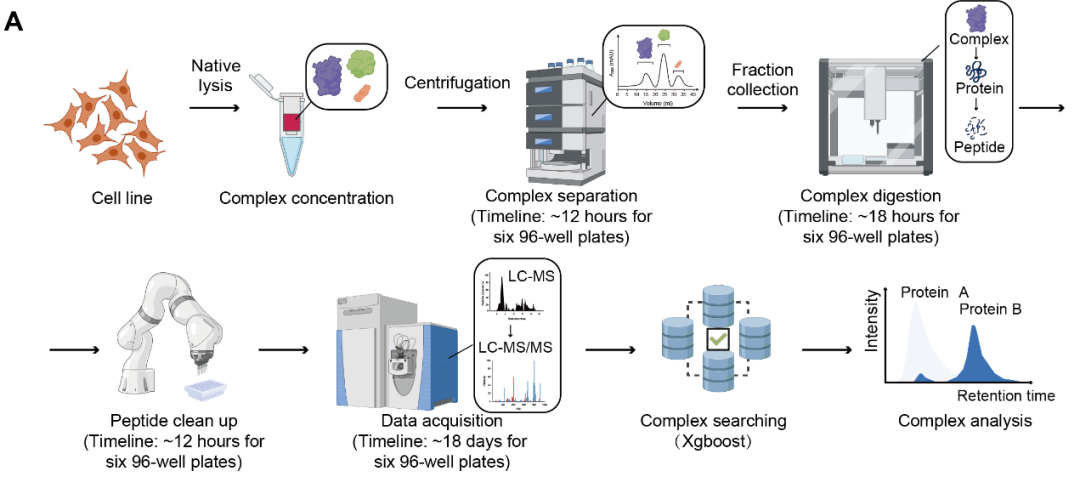
图2 ProteoAutoNet的基本流程
2)评估自动化机械臂实验平台的样品处理性能
研究团队通过同步处理三个细胞系(正常甲状腺细胞Nthy-ori 3-1,人甲状腺滤泡癌细胞FTC238,人甲状腺乳头状癌细胞TPC-1)的9个未分馏混合样本和540个分馏样本,对实验平台的蛋白质鉴定覆盖度与重复性进行评估(图3)。
对三个细胞系分别进行三次生物学重复的共分馏实验(图3B),并且鉴定每个细胞系内三次重复实验的蛋白质。最终,三种细胞系的生物学重复样本分别鉴定到2897、2938和2986个共有蛋白质,细胞系内三个生物学重复之间的蛋白质鉴定Jaccard相似系数均高于0.84,表明平台具有高度一致性(图3C)。蛋白质丰度在三个生物学重复样本之间表现出高相关性,Spearman相关系数均超过0.98(图3D)。
研究团队进一步将自动化实验平台与人工处理进行直接对比。以未分馏的TPC-1样本为例,自动化处理组的蛋白质数量变异系数(CV)由人工处理的蛋白质数量CV相比,从1.08%降至1.01%(图3E)。两组在蛋白质鉴定数量上高度一致:自动化处理为3408±34,人工处理为3404±37(图3B)。定量重复性分析显示,自动化处理组的中位CV更低(自动化组20.1% vs. 人工组21.6%),且四分位距更窄(自动化组12.6% vs. 人工组14.4%),体现出更稳定的定量性能(图3F)。
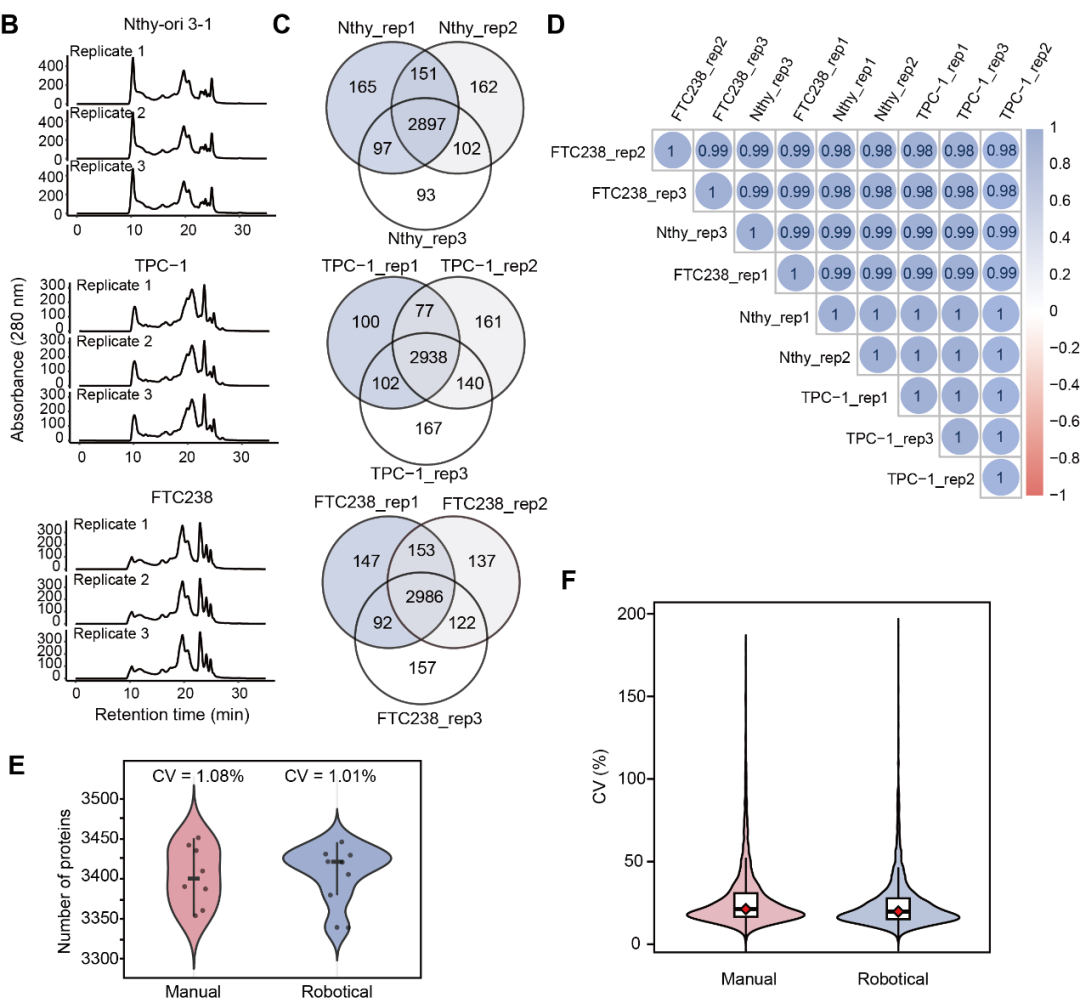
图3 ProteoAutoNet实验平台样品处理性能评估
2.XGBoost机器学习模型:教AI识别蛋白质的“朋友关系”
针对CF-MS数据的正样本(即已知真实PPIs)稀疏的问题,研究团队引入数据增强策略,通过对可靠的PPIs所产生的“共流出轨迹”数据进行随机扰动,扩增并多样化训练数据(图4),最终基于XGBoost构建预测模型。
在交叉验证中,模型取得AUROC = 0.81、AUPRC = 0.82的性能。在最优超参数条件下,内部验证集的AUROC和AUPRC分别为0.77和0.78。内部测试集分别达到0.78和0.79。在外部数据集,即FTC133细胞系中,以原始数据的天然类别比例进行验证,模型取得AUROC = 0.68。该性能下降反映不同细胞系间的差异以及天然蛋白质组数据中固有的类别不平衡问题。
在严格概率阈值(0.95)下的高置信度预测中,FTC133数据集的加权精度达到0.5。在FTC133细胞系鉴定到的18,811个高置信度PPIs中,有6558个是已有文献或数据库记录的PPIs。进一步分析显示,三个甲状腺细胞系(Nthy-ori 3-1,TPC-1,FTC238)共同鉴定的25,173个高置信度PPIs中,预测概率均高于0.95,其中1197个PPIs此前已有文献或数据库记录。
值得注意的是,现有PPIs预测方法通常需要整合多个数据集才能获得稳定性能,而基于单一数据集的AUROC通常仅约0.65。通过系统性结合数据增强与XGBoost算法,我们在保持天然类别不平衡的前提下,将单数据集的预测准确率提升了21.5%。
如果用一个更直观的比喻来理解这一过程:这就像在教AI识别社交网络中的朋友关系。ProteoAutoNet实验平台提供的,是大量蛋白质在不同条件下的“共流出轨迹”。已知的PPIs数据库,相当于已经确认的“好友关系”。数据增强策略,则是在不违背已知规律的前提下,对已知“好友”的“性格”、“价值观”指标进行随机轻微扰动。这促使模型的学习目标,从仅记忆训练数据中有限的、固定的“好友性格-价值观匹配”模式,转向理解“何种模式可能促成友谊”的通用规则,从而获得在新环境中判断潜在促成好友的能力。这正是模型能够在单一数据集上仍保持良好泛化能力的关键原因。
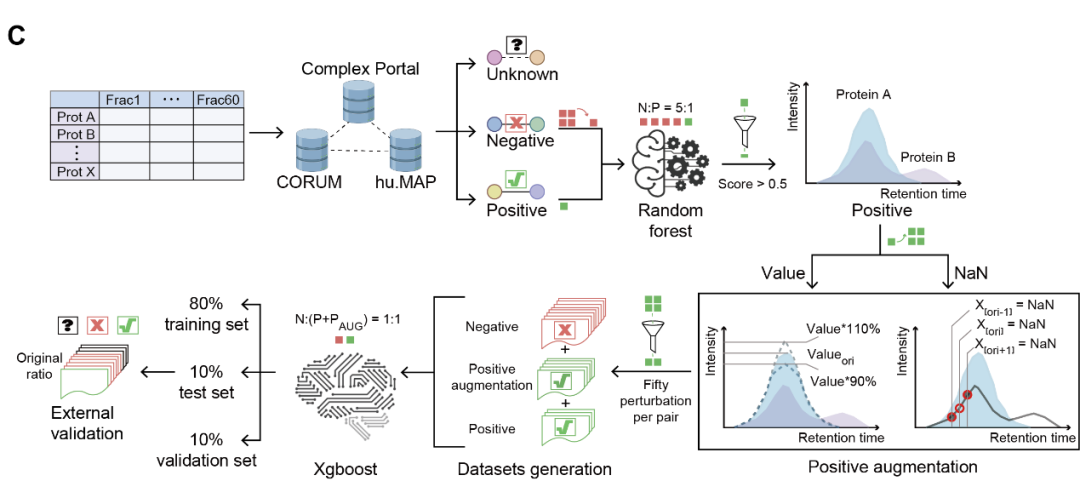
图4 ProteoAutoNet数据处理流程
将ProteoAutoNet应用于人正常甲状腺细胞(Nthy-ori 3-1)、人甲状腺滤泡癌细胞(FTC238)、人甲状腺乳头状癌细胞(TPC-1)后,研究团队共鉴定出25,173个高置信度PPIs。
首先,在三种细胞系中均检测到核糖体、蛋白酶体等保守的蛋白复合物。值得关注的是,在具有肺转移特性的甲状腺癌细胞系FTC238中,蛋白酶体与Prefoldin分子伴侣复合物显著上调(图5A, 5B),提示其可能参与肿瘤转移相关的蛋白质稳态调控。
此外,研究团队预测并验证一对此前未被实验报道的PPI:己糖激酶 1(HK1)与转谷氨酰胺酶 2(TGM2)在TPC-1细胞中呈现协同增强的蛋白丰度信号(图5C)。HK1是糖酵解起始步骤的关键酶,而TGM2已被证明与肿瘤侵袭和转移密切相关。该结果提示乳头状甲状腺癌的侵袭性可能与代谢重编程过程存在直接耦联。AlphaFold3的结构预测进一步为该互作提供了中高置信度的结构支持(图5D)。
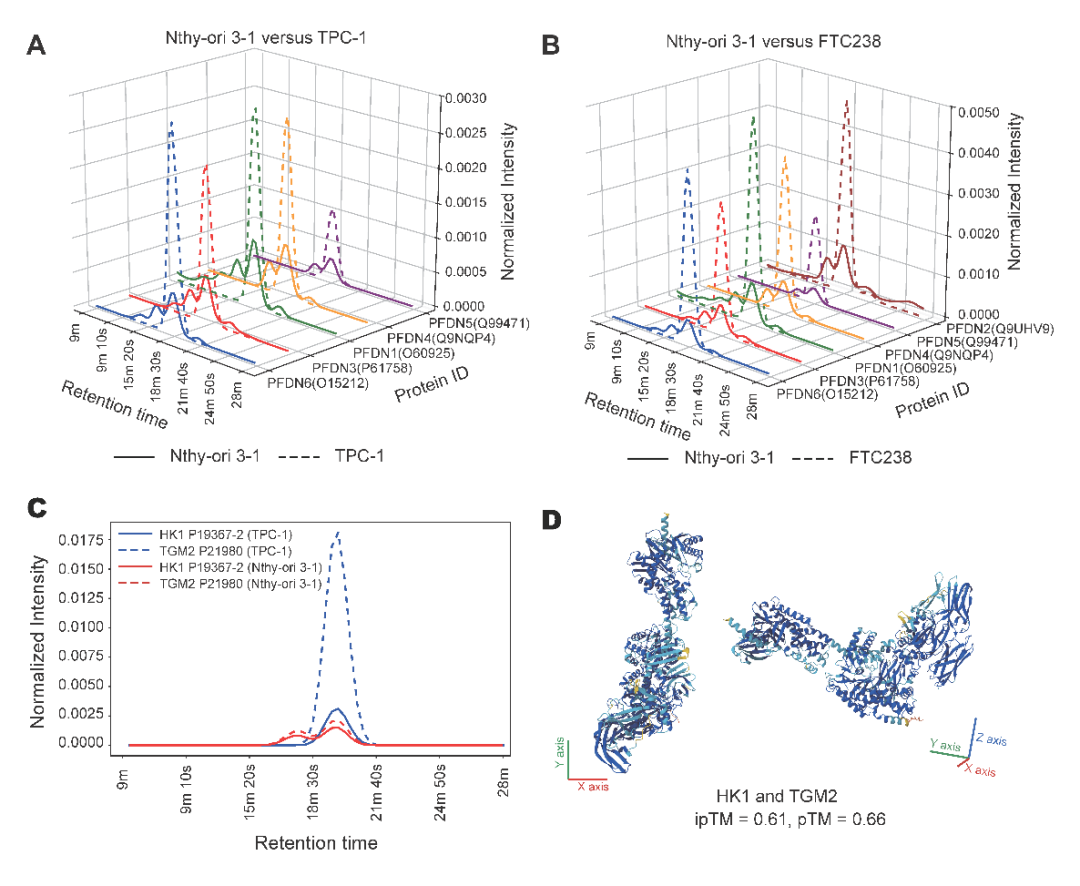
图5 ProteoAutoNet发现甲状腺癌细胞系中关键蛋白质相互作用
ProteoAutoNet展示了一种可能的未来方向:当自动化实验平台与机器学习模型真正协同工作,CF-MS研究有望成为一种高通量、可扩展、可泛化的蛋白质复合物研究工具。
原西湖大学医学院博士生吕梦葛为该研究第一作者。西湖大学医学院长聘副教授郭天南、助理研究员孙瑞、西湖实验室高级项目专家陈义为本研究共同通讯作者。山东第一医科大学医学信息与人工智能学院李相清副教授团队为项目搭建了Opentrons自动移液系统与JAKA机械臂的CF-MS前处理自动化平台,研究得到国家自然科学基金、国家重点研发计划、浙江省自然科学基金的支持。
西湖大学蛋白质组复杂科学实验室(Guomics)是依托西湖大学、西湖实验室(生命科学和生物医学浙江省实验室)和医学蛋白质组全国重点实验室成立的一个多学科交叉的AI蛋白质组学实验室。实验室长期从事蛋白质组学相关研究,联合人工智能,构建虚拟细胞,解析生物过程的原理,助力疾病诊疗。团队诚邀有志于虚拟细胞研究的优秀本科生、研究生及博士后研究人员加盟!实验室官网:guomics.com














